ml contains the code base to process glycan for machine learning, construct state-of-the-art machine learning models, train them, and analyze trained models + glycan representations. It currently contains the following modules:
model_training contains functions for training machine learning models
models describes some examples for machine learning architectures applicable to glycans
processing contains helper functions to prepare glycan data for model training
inference can be used to analyze trained models, make predictions, or obtain glycan representations
train_test_split contains various data split functions to get appropriate training and test sets
model_training
contains functions for training machine learning models
EarlyStopping
def EarlyStopping( patience:int=7, # epochs to wait after last improvement verbose:bool=False, # whether to print messages)->None:
Early stops the training if validation loss doesn’t improve after a given patience
train_model
def train_model( model:Module, # graph neural network for analyzing glycans dataloaders:dict, # dict with 'train' and 'val' loaders criterion:Module, # PyTorch loss function optimizer:Optimizer, # PyTorch optimizer, has to be SAM if mode != "regression" scheduler:LRScheduler, # PyTorch learning rate decay num_epochs:int=25, # number of epochs for training patience:int=50, # epochs without improvement until early stop mode:str='classification', # 'classification', 'multilabel', or 'regression' mode2:str='multi', # 'multi' or 'binary' classification return_metrics:bool=False, # whether to return metrics)->torch.nn.modules.module.Module |tuple[torch.nn.modules.module.Module, dict[str, dict[str, list[float]]]]:
trains a deep learning model on predicting glycan properties
training_setup
def training_setup( model:Module, # graph neural network for analyzing glycans lr:float, # learning rate lr_patience:int=4, # epochs before reducing learning rate factor:float=0.2, # factor to multiply lr on reduction weight_decay:float=0.0001, # regularization parameter mode:str='multiclass', # type of prediction task num_classes:int=2, # number of classes for classification gsam_alpha:float=0.0, # if >0, uses GSAM instead of SAM optimizer warmup_epochs:int=5, # if >0, uses a learning rate warm-up schedule for training stability)->tuple:
prepares optimizer, learning rate scheduler, and loss criterion for model training
train_ml_model
def train_ml_model( X_train:pandas.DataFrame |list, # training data/glycans X_test:pandas.DataFrame |list, # test data/glycans y_train:list, # training labels y_test:list, # test labels mode:str='classification', # 'classification' or 'regression' feature_calc:bool=False, # calculate motifs from glycans return_features:bool=False, # return calculated features feature_set:list=['known', 'exhaustive'], # feature set for annotations additional_features_train:pandas.DataFrame |None=None, # additional training features additional_features_test:pandas.DataFrame |None=None, # additional test features)->xgboost.sklearn.XGBModel |tuple[xgboost.sklearn.XGBModel, pandas.DataFrame, pandas.DataFrame]:
wrapper function to train standard machine learning models on glycans
human = [1if k =='Homo_sapiens'else0for k in df_species[df_species.Order=='Primates'].Species.values.tolist()]X_train, X_test, y_train, y_test = general_split(df_species[df_species.Order=='Primates'].glycan.values.tolist(), human)model_ft, _, X_test = train_ml_model(X_train, X_test, y_train, y_test, feature_calc =True, feature_set = ['terminal'], return_features =True)
Calculating Glycan Features...
Training model...
Evaluating model...
Accuracy of trained model on separate validation set: 0.8578856152512998
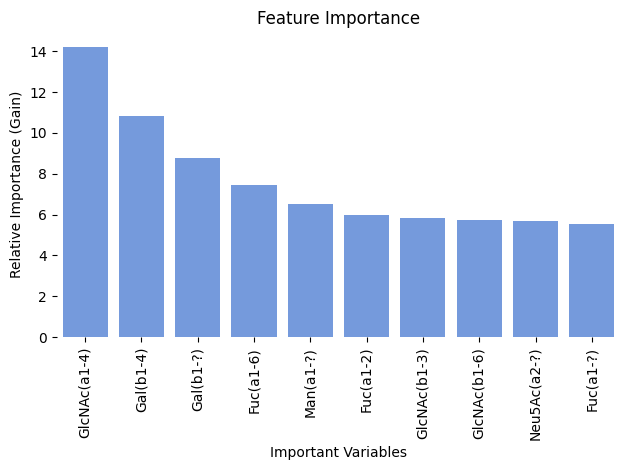
analyze_ml_model
def analyze_ml_model( model:XGBModel, # trained ML model from train_ml_model)->None:
plots relevant features for model prediction
analyze_ml_model(model_ft)
get_mismatch
def get_mismatch( model:XGBModel, # trained ML model from train_ml_model X_test:DataFrame, # motif dataframe for validation y_test:list, # test labels n:int=10, # number of returned misclassifications)->list: # misclassifications and predicted probabilities
analyzes misclassifications of trained machine learning model
describes some examples for machine learning architectures applicable to glycans. The main portal is prep_models which allows users to setup (trained) models by their string names
SweetNet
def SweetNet( lib_size:int, # number of unique tokens for graph nodes num_classes:int=1, # number of output classes (>1 for multilabel) hidden_dim:int=128, # dimension of hidden layers)->None:
Base class for all neural network modules.
Your models should also subclass this class.
Modules can also contain other Modules, allowing them to be nested in a tree structure. You can assign the submodules as regular attributes::
import torch.nn as nn
import torch.nn.functional as F
class Model(nn.Module):
def __init__(self) -> None:
super().__init__()
self.conv1 = nn.Conv2d(1, 20, 5)
self.conv2 = nn.Conv2d(20, 20, 5)
def forward(self, x):
x = F.relu(self.conv1(x))
return F.relu(self.conv2(x))
Submodules assigned in this way will be registered, and will also have their parameters converted when you call :meth:to, etc.
.. note:: As per the example above, an __init__() call to the parent class must be made before assignment on the child.
:ivar training: Boolean represents whether this module is in training or evaluation mode. :vartype training: bool
LectinOracle
def LectinOracle( input_size_glyco:int, # number of unique tokens for graph nodes hidden_size:int=128, # layer size for graph convolutions num_classes:int=1, # number of output classes (>1 for multilabel) data_min:float=-11.355, # minimum observed value in training data data_max:float=23.892, # maximum observed value in training data input_size_prot:int=960, # dimensionality of protein representations)->None:
Base class for all neural network modules.
Your models should also subclass this class.
Modules can also contain other Modules, allowing them to be nested in a tree structure. You can assign the submodules as regular attributes::
import torch.nn as nn
import torch.nn.functional as F
class Model(nn.Module):
def __init__(self) -> None:
super().__init__()
self.conv1 = nn.Conv2d(1, 20, 5)
self.conv2 = nn.Conv2d(20, 20, 5)
def forward(self, x):
x = F.relu(self.conv1(x))
return F.relu(self.conv2(x))
Submodules assigned in this way will be registered, and will also have their parameters converted when you call :meth:to, etc.
.. note:: As per the example above, an __init__() call to the parent class must be made before assignment on the child.
:ivar training: Boolean represents whether this module is in training or evaluation mode. :vartype training: bool
LectinOracle_flex
def LectinOracle_flex( input_size_glyco:int, # number of unique tokens for graph nodes hidden_size:int=128, # layer size for graph convolutions num_classes:int=1, # number of output classes (>1 for multilabel) data_min:float=-11.355, # minimum observed value in training data data_max:float=23.892, # maximum observed value in training data input_size_prot:int=1000, # maximum protein sequence length for padding/cutting)->None:
Base class for all neural network modules.
Your models should also subclass this class.
Modules can also contain other Modules, allowing them to be nested in a tree structure. You can assign the submodules as regular attributes::
import torch.nn as nn
import torch.nn.functional as F
class Model(nn.Module):
def __init__(self) -> None:
super().__init__()
self.conv1 = nn.Conv2d(1, 20, 5)
self.conv2 = nn.Conv2d(20, 20, 5)
def forward(self, x):
x = F.relu(self.conv1(x))
return F.relu(self.conv2(x))
Submodules assigned in this way will be registered, and will also have their parameters converted when you call :meth:to, etc.
.. note:: As per the example above, an __init__() call to the parent class must be made before assignment on the child.
:ivar training: Boolean represents whether this module is in training or evaluation mode. :vartype training: bool
NSequonPred
def NSequonPred()->None:
Base class for all neural network modules.
Your models should also subclass this class.
Modules can also contain other Modules, allowing them to be nested in a tree structure. You can assign the submodules as regular attributes::
import torch.nn as nn
import torch.nn.functional as F
class Model(nn.Module):
def __init__(self) -> None:
super().__init__()
self.conv1 = nn.Conv2d(1, 20, 5)
self.conv2 = nn.Conv2d(20, 20, 5)
def forward(self, x):
x = F.relu(self.conv1(x))
return F.relu(self.conv2(x))
Submodules assigned in this way will be registered, and will also have their parameters converted when you call :meth:to, etc.
.. note:: As per the example above, an __init__() call to the parent class must be made before assignment on the child.
:ivar training: Boolean represents whether this module is in training or evaluation mode. :vartype training: bool
init_weights
def init_weights( model:Module, # neural network for analyzing glycans mode:str='sparse', # initialization algorithm: 'sparse', 'kaiming', 'xavier' sparsity:float=0.1, # proportion of sparsity after initialization)->None:
initializes linear layers of PyTorch model with a weight initialization
prep_model
def prep_model( model_type:Literal, # type of model to create num_classes:int, # number of unique classes for classification libr:dict[str, int] |None=None, # dictionary of form glycoletter:index trained:bool=False, # whether to use pretrained model hidden_dim:int=128, # hidden dimension for the model (SweetNet only))->Module: # initialized PyTorch model
wrapper to instantiate model, initialize it, and put it on the GPU
processing
contains helper functions to prepare glycan data for model training
dataset_to_graphs
def dataset_to_graphs( glycan_list:list, # list of IUPAC-condensed glycan sequences labels:list, # list of labels libr:dict[str, int] |None=None, # dictionary of glycoletter:index label_type:dtype=torch.int64, # tensor type for label)->list: # list of node/edge/label data tuples
wrapper function to convert a whole list of glycans into a graph dataset
def dataset_to_dataloader( glycan_list:list, # list of IUPAC-condensed glycans labels:list, # list of labels libr:dict[str, int] |None=None, # dictionary of glycoletter:index batch_size:int=32, # samples per batch shuffle:bool=True, # shuffle samples in dataloader drop_last:bool=False, # drop last batch extra_feature:list[float] |None=None, # additional input features label_type:dtype=torch.int64, # tensor type for label augment_prob:float=0.0, # probability of data augmentation generalization_prob:float=0.2, # probability of wildcarding)->DataLoader: # dataloader for training
wrapper function to convert glycans and labels to a torch_geometric DataLoader
def split_data_to_train( glycan_list_train:list, # training glycans glycan_list_val:list, # validation glycans labels_train:list, # training labels labels_val:list, # validation labels libr:dict[str, int] |None=None, # dictionary of glycoletter:index batch_size:int=32, # samples per batch drop_last:bool=False, # drop last batch extra_feature_train:list[float] |None=None, # additional training features extra_feature_val:list[float] |None=None, # additional validation features label_type:dtype=torch.int64, # tensor type for label augment_prob:float=0.0, # probability of data augmentation generalization_prob:float=0.2, # probability of wildcarding)->dict: # dictionary of train/val dataloaders
wrapper function to convert split training/test data into dictionary of dataloaders
>can be used to analyze trained models, make predictions, or obtain glycan representations
glycans_to_emb
def glycans_to_emb( glycans:list, # list of glycans in IUPAC-condensed model:Module, # trained graph neural network for analyzing glycans libr:dict[str, int] |None=None, # dictionary of form glycoletter:index batch_size:int=32, # batch size used during training rep:bool=True, # True returns representations, False returns predicted labels class_list:list[str] |None=None, # list of unique classes to map predictions multilabel:bool=False, # whether to output predictions for a multilabel-task)->pandas.DataFrame |list[str]: # dataframe of representations or list of predictions
Returns a dataframe of learned representations for a list of glycans
get_lectin_preds
def get_lectin_preds( prot:str, # protein amino acid sequence glycans:list, # list of glycans in IUPAC-condensed model:Module, # trained LectinOracle-type model prot_dic:dict[str, list[float]] |None=None, # dict of protein sequence:ESMC representation background_correction:bool=False, # whether to correct predictions for background correction_df:pandas.DataFrame |None=None, # background prediction for glycans batch_size:int=128, # batch size used during training libr:dict[str, int] |None=None, # dict of glycoletter:index sort:bool=True, # whether to sort prediction results descendingly flex:bool=False, # LectinOracle (False) or LectinOracle_flex (True))->DataFrame: # glycan sequences and predicted binding
Wrapper that uses LectinOracle-type model for predicting binding of protein to glycans
get_Nsequon_preds
def get_Nsequon_preds( prots:list, # 20 AA + N + 20 AA sequences; replace missing with 'z' model:Module, # trained NSequonPred-type model prot_dic:dict, # dict of protein sequence:ESM1b representation)->DataFrame: # protein sequences and predicted likelihood
Predicts whether an N-sequon will be glycosylated
get_esmc_representations
def get_esmc_representations( prots:list, # list of protein sequences to convert model:Module, # trained ESMC model)->dict: # dict of protein sequence:ESMC-300M representation
Retrieves ESMC-300M representations of protein for using them as input for LectinOracle
In order to run get_esmc_representations, you first have to run this snippet:
contains various data split functions to get appropriate training and test sets
hierarchy_filter
def hierarchy_filter( df_in:DataFrame, # dataframe of glycan sequences and taxonomic labels rank:str='Domain', # taxonomic rank to filter min_seq:int=5, # minimum glycans per class wildcard_seed:bool=False, # seed wildcard glycoletters wildcard_list:list[str] |None=None, # glycoletters for wildcard wildcard_name:str|None=None, # wildcard name in IUPAC r:float=0.1, # replacement rate col:str='glycan', # column name for glycans)->tuple: # train/val splits and mappings
stratified data split in train/test at the taxonomic level, removing duplicate glycans and infrequent classes
def general_split( glycans:list, # list of IUPAC-condensed glycans labels:list, # list of prediction labels test_size:float=0.2, # size of test set)->tuple: # train/test splits
def prepare_multilabel( df:DataFrame, # dataframe with one glycan-association per row rank:str='Species', # label column to use glycan_col:str='glycan', # column with glycan sequences)->tuple: # unique glycans and their label vectors
converts a one row per glycan-species/tissue/disease association file to a format of one glycan - all associations